 Introduction
Introduction
Innovation is a main driver of every industry. This is especially true for medical device companies which, in conjunction with surgeons, are inventing, developing, and commercializing technologies to improve clinical outcomes for patients worldwide. With over 6,500 medical device companies in the United States alone, the global device market is expected to reach $543.9 billion by 2020. This growth, driven by an aging population, increasing healthcare expenditure and technology advancement, will drive the Food and Drug Administration (FDA) to review an ever-increasing number of device applications.
 The speed with which a new medical device can be brought to market is greatly influenced by the resources available to the FDA. As such, the FY2018 Presidential Budget Request (PBR) aims to continue the trajectory of innovation by increasing the number of reviewers, in order to streamline the process of submission review. It attempts to accomplish this task through a restructuring of the funding sources for reviewers while maintaining the same threshold of review quality and least burdensome practices.
The speed with which a new medical device can be brought to market is greatly influenced by the resources available to the FDA. As such, the FY2018 Presidential Budget Request (PBR) aims to continue the trajectory of innovation by increasing the number of reviewers, in order to streamline the process of submission review. It attempts to accomplish this task through a restructuring of the funding sources for reviewers while maintaining the same threshold of review quality and least burdensome practices.
Analysis by Musculoskeletal Clinical Regulatory Advisers, LLC (MCRA) of the FY2018 PBR indicates that, if passed, the FDA budget could hurt the capacity of the FDA to review product applications; including Premarket Approval Applications (PMAs), 510(k)s and de Novo submissions, among others. PMAs usually represent the regulatory pathway for the most innovative of technologies, and are the base fee that other user fees are based upon. Fees for 510(k)s, PMA supplements, and other regulatory submissions are determined as a percentage of the base PMA fee. By cutting the budget authority for the Medical Device Review Program, experts at MCRA are concerned that review timelines could lengthen, the quality of reviews may decrease, and the communication between the FDA and industry might become more difficult. As a result of this dynamic, the regulatory and financial barriers for companies will increase and create market uncertainty in the future.
The Budget Request
The FDA Center for Devices and Radiological Health (CDRH) is responsible for ensuring the safety and effectiveness of medical devices through the Medical Devices Review Program. CDRH has a number of regulatory pathways such as the de Novo process to the 510(k) and PMA pathways, in order to diversify the avenues of innovation for industry. This resulted in a variety of submissions being reviewed by the FDA and diverse, innovative products reaching the market. Since 2012, CDRH established target goals for improving the review times of PMAs and 510(k)s. According to the FY2016 Performance Report from CDRH, 100% of all PMAs and 98% of all 510(k) submissions received a decision within established goals of 390 days and 130 days, respectively. This is an improvement from 2013 when 84% of PMAs and 95% of 510(k)s met their review time-frame goals. However, this continued performance improvement hinges on reliable federal funding of the FDA.
The FDA is primarily funded by a budget appropriated through Congress. This budget is supplemented by user fees paid by companies submitting product applications, such as PMAs or 510(k)s. For medical devices, these fees are determined by the Medical Device User Fee and Modernization Act of 2002 (MDUFMA) and are reauthorized and updated every five years in the Medical Device User Fee Amendments (MDUFA). The user fees for fiscal years 2013 to 2017 were set in 2012 by MDUFA III and cover the associated costs of FDA reviewer salary and benefits. The next generation of user fees, MDUFA IV, needs to be updated and approved by Congress by September 30, 2017. The future of this fee based program hinges on this Congressional action.
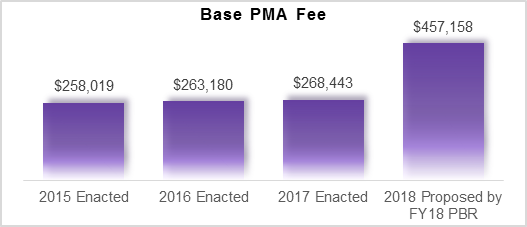 The PBR proposes the amount of funding recommended for each part of the federal government for the 2018 fiscal year, which begins on October 1, 2017. The PBR recommendation is to reduce the FDA budget authority by $870 million (31.6%). This includes a $243 million cut to the Medical Device Review Program. This would mean that the entire budget authority of the program would be cut. The PBR proposes making up the funding through increasing user fee revenue. However, unless there is a dramatic increase in the volume of regulatory submissions to FDA, the Administration and Congress will need to increase user fees by 70.3%. This would need to be done by amending MDUFA IV or changing the user fees through another authorization.
The PBR proposes the amount of funding recommended for each part of the federal government for the 2018 fiscal year, which begins on October 1, 2017. The PBR recommendation is to reduce the FDA budget authority by $870 million (31.6%). This includes a $243 million cut to the Medical Device Review Program. This would mean that the entire budget authority of the program would be cut. The PBR proposes making up the funding through increasing user fee revenue. However, unless there is a dramatic increase in the volume of regulatory submissions to FDA, the Administration and Congress will need to increase user fees by 70.3%. This would need to be done by amending MDUFA IV or changing the user fees through another authorization.
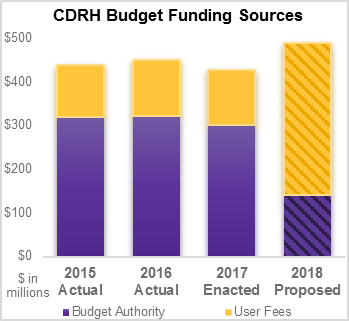 These budget changes could impact the performance and capabilities of the program to maintain current performance levels.
These budget changes could impact the performance and capabilities of the program to maintain current performance levels.
Impact on industry
The current FDA funding structure was designed to ensure a constant level of funding even during low-submission periods. While the proposed budget cuts are not expected to impact staffing levels for product reviewers, there may be other consequences. Many experts anticipate an increase in review times and that a negative impact on the quality of new product reviews may result in higher regulatory hurdles for submitting companies to overcome.
According to Glenn Stiegman, Senior Vice President of Regulatory and Clinical Affairs at MCRA and former Branch Chief of Orthopedics at the FDA, “By shifting to a budget which relies substantially or entirely on user fees, and at the same time proposing a significant increase to user fees for all medical device submissions, this may have a negative impact on innovation. Companies already find it expensive and challenging to conduct a successful clinical study and take the higher risk pathway of a PMA. Raising user fees may deter companies from taking the time and cost risks, and not go through the process of marketing innovative products in the US. Additionally, 510(k)s and de Novo will also become less attractive, as their costs continue to rise. It is important to be aware that if companies choose to avoid the PMA pathway, the FDA’s funding from user fees will decrease, and we could see a depletion in review staff. A review of a PMA is complex and time-consuming, and if less resources are available to the FDA to assist with the review or even lead the review, then timelines will not be met. Currently companies are able to commercialize new technologies in the US within a known and expected timeline when the FDA has the right staff and right funding. This budgetary change could reverse that trend.”
Conclusion
User fees were designed to accelerate the process of regulatory submission review. With the proposed PBR aimed to fund product review programs exclusively through user fees, review times may increase and make it longer for new medical devices to reach the commercial markets. Therefore, it is critical to develop an integrated clinical and regulatory strategy that presents the appropriate data to FDA as effectively and efficiently as possible.
As of July 17th, the House of Representatives passed the FDA Reauthorization Act (H.R. 2430), which includes MUDFA IV. The Senate has placed the bill on the Legislative Calendar, despite the Administration urging “Congress to provide for 100 percent user fee funding within the [review programs].” The support for this bill will be indicative of whether the Congress is open or not to the FDA funding source changes proposed in the PBR.
Most companies lack access to former FDA regulators to interpret the evolving regulatory requirements and to guide them through the complex process of product review and approval. Whether or not the PBR passes, significant changes are on the horizon. There is a movement within the current Administration and Congress to fundamentally change the organization of and structure of the FDA. While the long term impact of such changes are uncertain, those medical device companies that leverage the expertise and experience of top level resources will likely be in the best position to achieve success.
- Department of Commerce. International Trade Administration. Medical Technology Spotlight. Washington, DC, 2017. N/A.
- Food and Drug Administration. Center for Devices and Radiological Health. FY2016 MDUFA Performance Report. Washington, DC, 2016. 13.
- FDA. CDRH. FY2013 MDUFA Performance Report. Washington, DC, 2013. 5.
- FDA. Office of the Commissioner. Justification of Estimates for Appropriations Committees. Washington, DC, 2017. 121.
- Executive Office of the President. Office of Management and Budget. Major Savings and Reforms: Budget of the U.S. Government Fiscal Year 2018. Washington, DC, 2017. 39.
- Executive Office of the President. Press Office. H.R. 2430 – FDA Reauthorization Act of 2017: Statement of Administration Policy. Washington, DC, 2017. N/A.
CREDIT: Whitepaper from MCRA